- Mutational signature patterns of of breast cancer tumors based on stage, grade and subtype
- Next-Gen sequence analysis, optimization of variant calling pipelines
- ECemble: An enzyme classification method to study the role of gut microbiome in human metabolism
- MetaID: Taxonomic profiling of metagenomic samples down to the strain level
- Graph mining and module detection in protein-protein interaction networks
- Computational methods for predicting protein subcellular localization
- A gene fingerprint-based algorithm for fusion genes detection from RNA-Seq short reads
- Functional characterization of healthy adult human brain and its application to study neurodevelopmental disorders
- Determining the clonal heterogeneity in tumors using exome sequencing data
- Integrative molecular characterization of glioblastoma
- Integrative data analysis of pancreatic tumor genomes
Mutational signature patterns of breast cancer tumors based on stage, grade and subtype
Breast cancers exhibit highly heterogeneous molecular profiles. Although gene expression profiles have been used to predict the risks and prognostic outcomes of breast cancers, the high variability of gene expression limits its clinical application. In contrast, genetic mutation profiles would be more advantageous than gene expression profiles because genetic mutations can be stably detected and the mutational heterogeneity widely exists in breast cancer genomes.
We analyzed 98 breast cancer whole exome samples that were sorted into three subtypes, two grades and two stages. The sum deleterious effect of all mutations in each gene was scored to identify differentially mutated genes (DMGs) for this case-control study. DMGs were corroborated using extensive published knowledge. Functional consequences of deleterious SNVs on protein structure and function were also investigated. Mutational profiling at gene- and SNV-level revealed differential patterns within each breast cancer comparison group, and the gene signatures correlate with expected prognostic characteristics of breast cancer classes. Some of the genes and SNVs identified in this study show high promise and are worthy of further investigation by experimental studies.
Click on image for larger version.
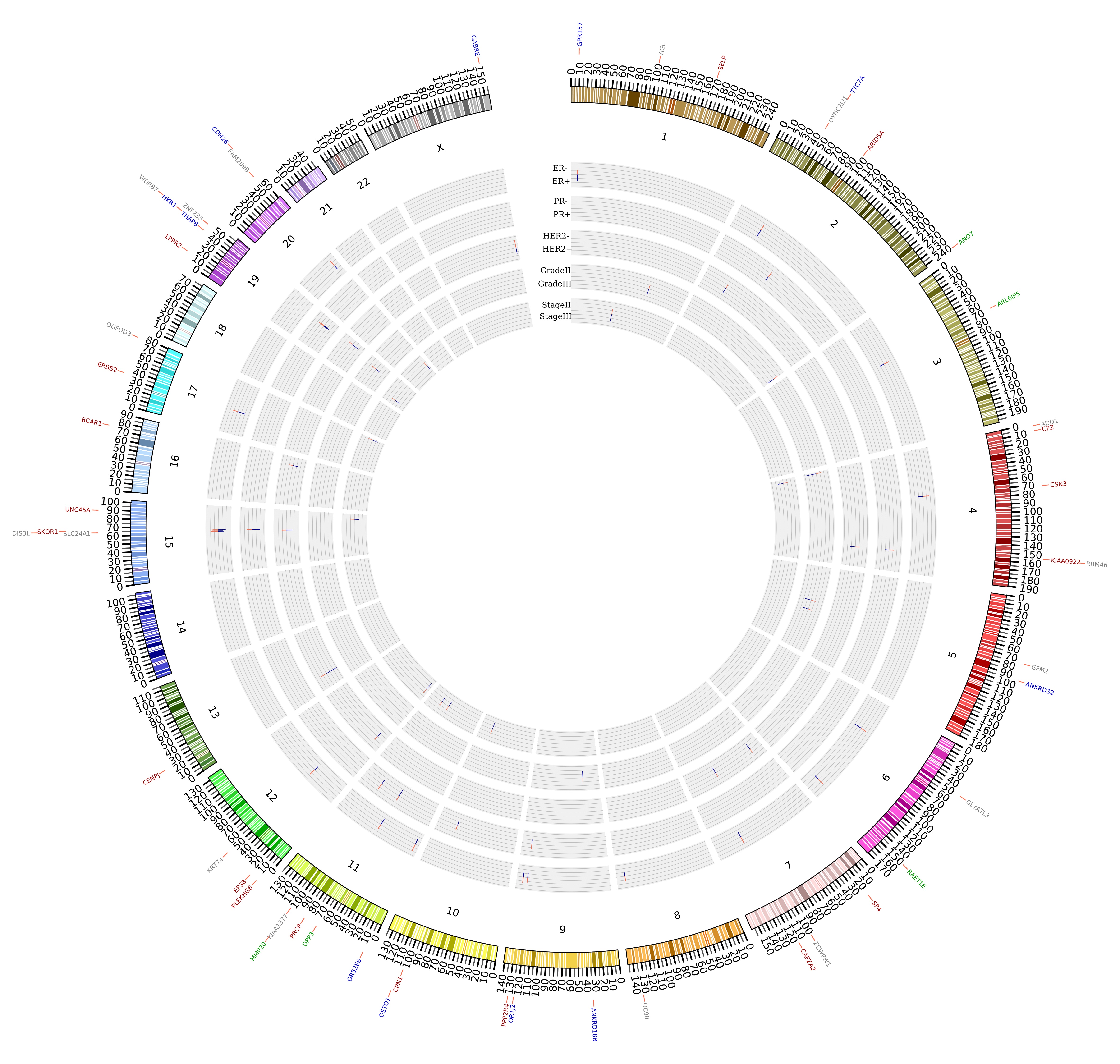 |
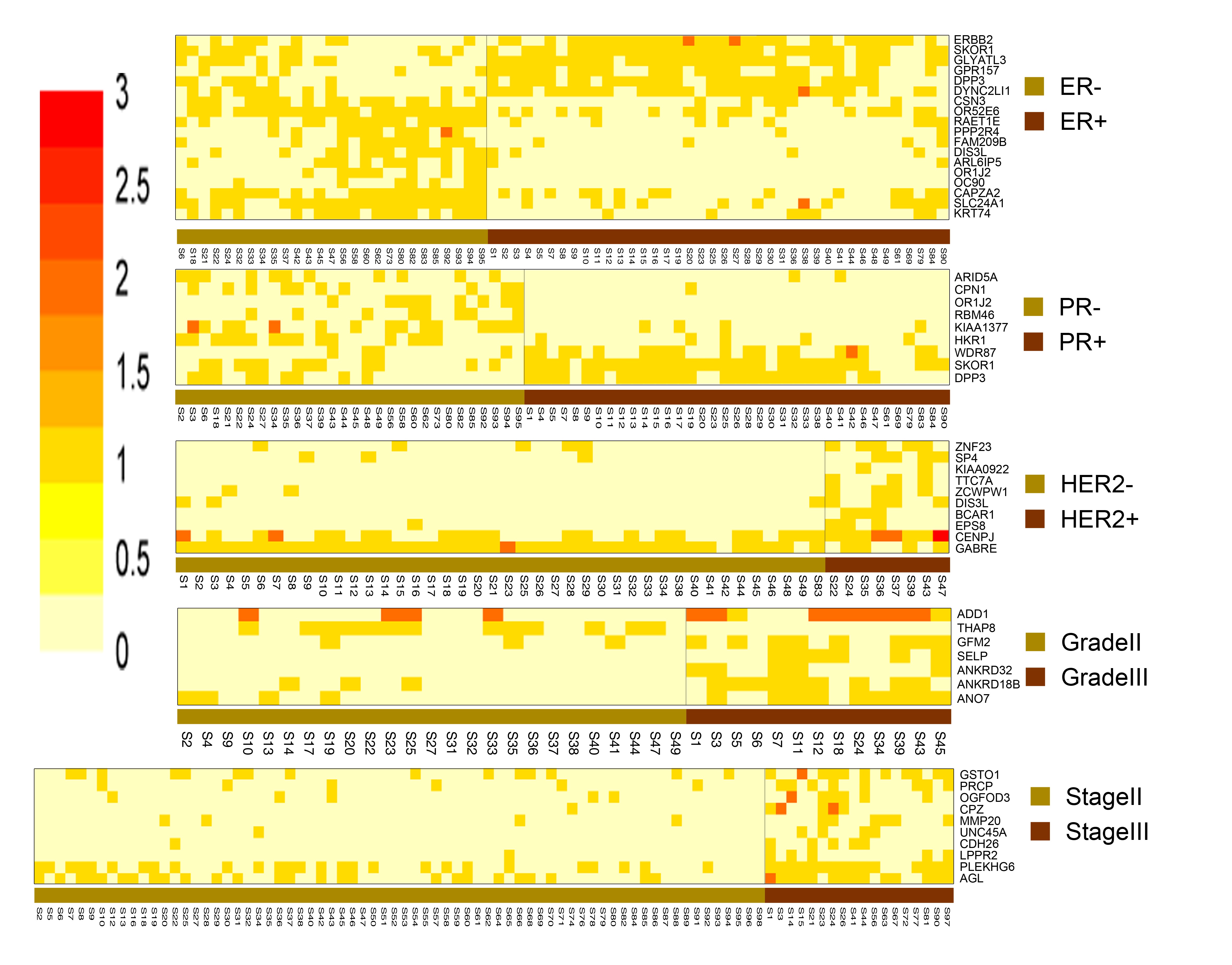 |
| The differentially mutated genes between breast cancer subtypes | The deleterious mutation scores for the differentially mutated genes across the compared samples |
Published articles related to this project:
- Li Y, Wang X, Vural S, Mishra N, Cowan KH, Guda C. Exome analysis reveals differentially mutated gene signatures of stage, grade and subtype in breast cancers. PLOS ONE (2015) 10(3):e0119383.
[PubMed]
Next-Gen sequence analysis, optimization of variant calling pipelines
High-throughput sequencing, especially exome sequencing, is fast becoming a popular diagnostic tool in the clinical setting, but it has become more and more difficult to determine which tools are the best at analyzing this sequencing data. Previously, researchers have had to use simulated data sets or gene chips as a form of validation to determine the best analysis pipeline, but both have drawbacks and biases. In this study we use the NIST Genome in a Bottle results as a novel method for exome analysis pipeline validation, which do not contain these inherent drawbacks.
Using the NIST Genome in a Bottle variant list as a golden standard, we were able to use six different aligners - Bowtie2, BWA mem, BWA sampe, CUSHAW3, MOSAIK, and Novoalign - and five different variant callers - FreeBayes, GATK HaplotypeCaller, GATK UnifiedGenotyper, SAMtools mpileup, and SNPSVM - to determine which pipeline performs the best on a standard human exome. SNVs were compared in all feasible pipelines by calculating sensitivities at different depths to determine which pipelines ultimately performed the best. We found that among the 30 different pipelines tested, Novoalign in conjunction with GATK UnifiedGenotyper exhibited the highest sensitivity while maintaining a low number of false positives for SNVs. However, it is readily apparent that indels are still difficult for any pipeline to handle with none of the tools achieving an average sensitivity higher than 33% or a Positive Predictive Value (PPV) higher than 53%. Additionally, this work highlights the fact that standard analysis pipelines are missing a large majority of the variants - both SNVs and indels - present in a normal human exome. Lastly, as expected, it was found that aligners can play as vital a role in variant detection as variant callers themselves.
Click on image for larger version.
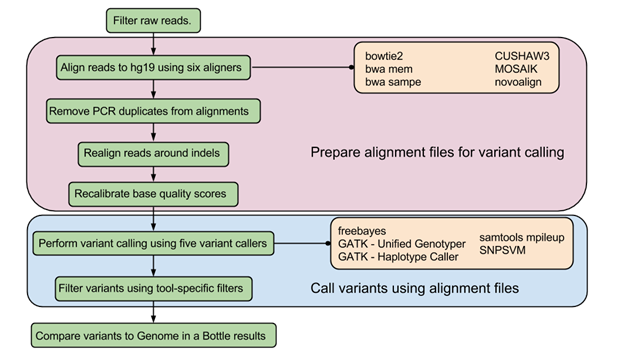 |
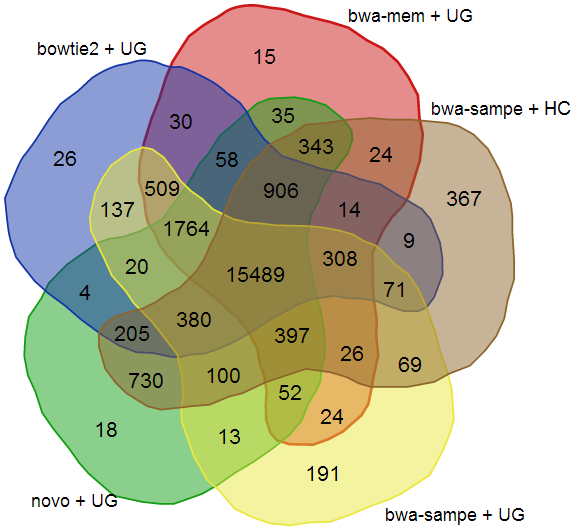 |
| Schematic of the data analysis pipeline used | The intersection of the SNVs identified by the top five pipelines |
Published articles related to this project:
- Cornish A, Guda C. A comparison of variant calling pipelines using Genome-in-a-Bottle as a reference. BioMed Research International (2015). [PubMed]
- Ozturk F, Li Y, Zhu X, Guda C, Nawshad A. Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFbeta3-knockout mice alleles: RNA-Seq analysis of TGFbeta3 Mice. BMC Genomics (2013) 14:113 [PubMed]
ECemble: An enzyme classification method to study the role of gut microbiome in human metabolism
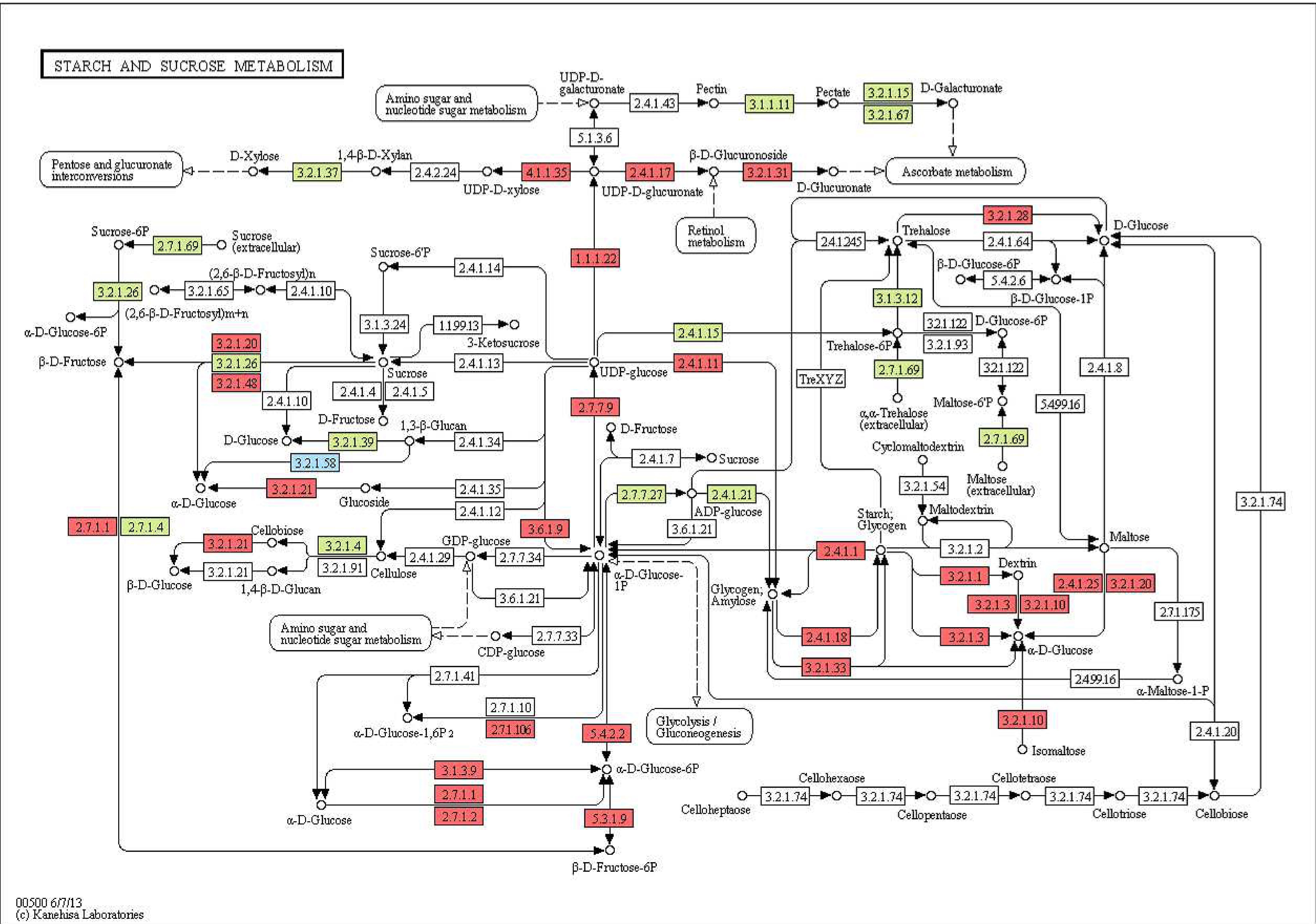
Enzymes encoded by the human gut microbiome play an essential role in the human metabolism. To annotate the full enzyme complements of species in the genomic and metagenomic projects, we developed a method called ECemble, to identify enzymes and enzyme classes and study the human gut metabolic pathways. ECemble method uses an ensemble of machine-learning methods to accurately model and predict enzymes from protein sequences and also identifies the enzyme classes and subclasses at the finest resolution. We applied ECemble to predict the entire complements of enzymes from ten sequenced proteomes including the human proteome. We also applied this method to predict enzymes encoded by the human gut microbiome from gut metagenomic samples, and to study the role played by the microbe-derived enzymes in the human metabolism. After mapping the known and predicted enzymes to canonical human pathways, we identified 48 pathways that have at least one bacteria-encoded enzyme, which demonstrates the complementary role of gut microbiome in human gut metabolism. These pathways are primarily involved in metabolizing dietary nutrients such as carbohydrates, amino acids, lipids, cofactors and vitamins. The ECemble method is able to hierarchically assign high quality enzyme annotations to genomic and metagenomic data. This study demonstrated the real application of ECemble to understand the indispensable role played by microbe-encoded enzymes in the healthy functioning of human metabolic systems.
Click on image for larger version.
 |
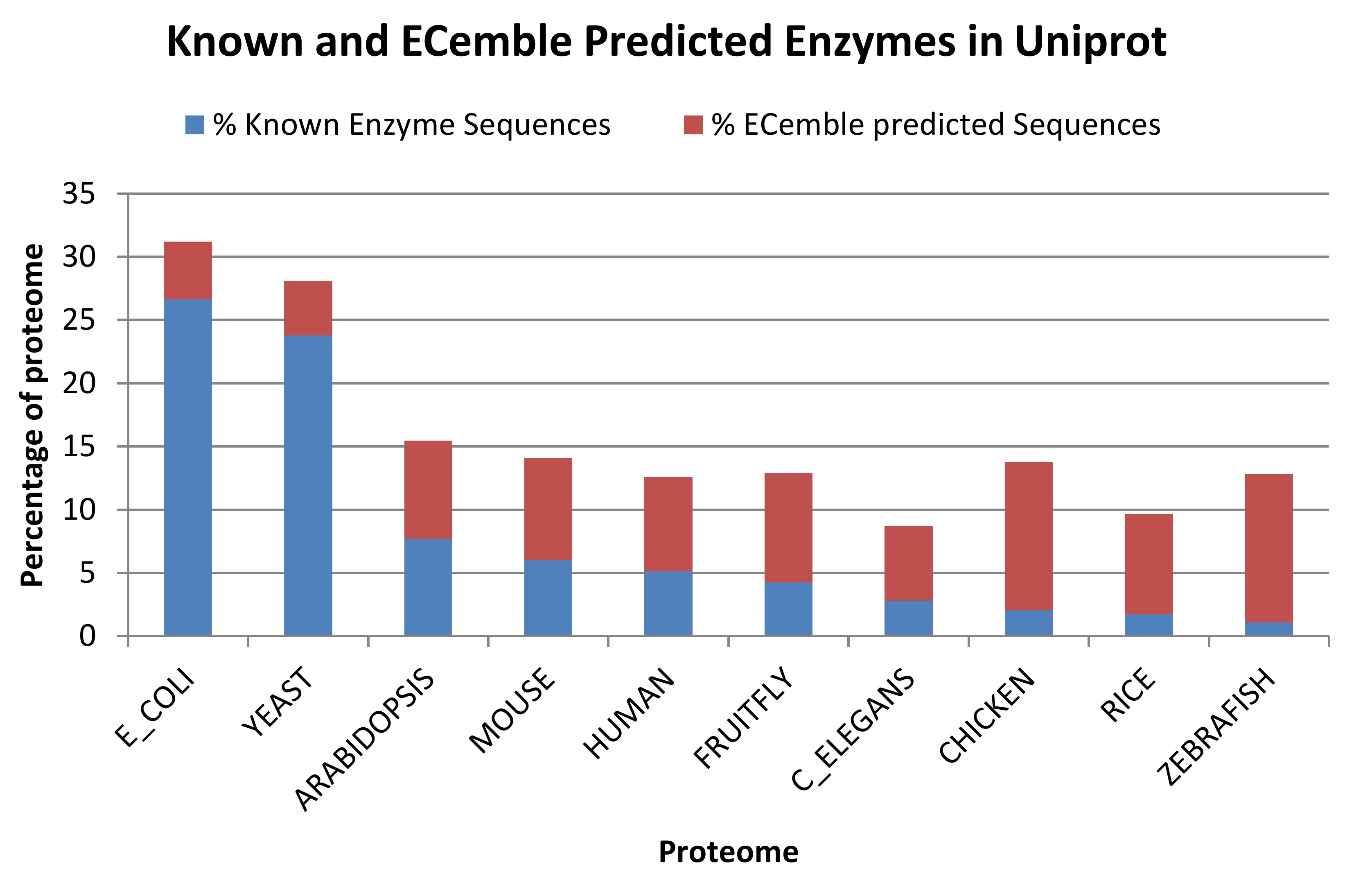 |
| Schematic representation of the ECemble method and its application | Fractions of known and ECemble predicted enzymes in the proteomes of 10 model organisms from UniProt |
 |
|
| Fructose and mannose metabolism pathway | |
Published articles related to this project:
- Mohammed A, Guda C. Application of a hierarchical enzyme classification method reveals the role of gut microbiome in human metabolism. BMC Genomics (2015) 16(Suppl 7):S16 doi:10.1186/1471-2164-16-S7-S16 [link]
MetaID: Taxonomic profiling of metagenomic samples down to the strain level
Several computational methods are available for taxonomic profiling at the genus- and species-level, but none of these methods are effective at the strain-level identification due to the increasing difficulty in detecting variation at that level. Here, we present MetaID, an alignment-free n-gram based approach that can accurately identify microorganisms at the strain level and estimate the abundance of each organism in a sample, given a metagenomic sequencing dataset.
MetaID is an n-gram based method that calculates the profile of unique and common n-grams from the dataset of 2,031 prokaryotic genomes and assigns weights to each n-gram using a scoring function. This scoring function assigns higher weightage to the n-grams that appear in fewer genomes and vice versa; thus, allows for effective use of both unique and common n-grams for species identification. The proposed scoring function and approach is able to accurately identify and estimate the entire taxa in any metagenomic community. The weights assigned to the common n-grams by our scoring function are precisely calibrated to match the reads up to the strain level. The generic approach employed in this method can be applied for accurate identification of a wide variety of microbial species (viruses, prokaryotes and eukaryotes) present in any environmental sample.
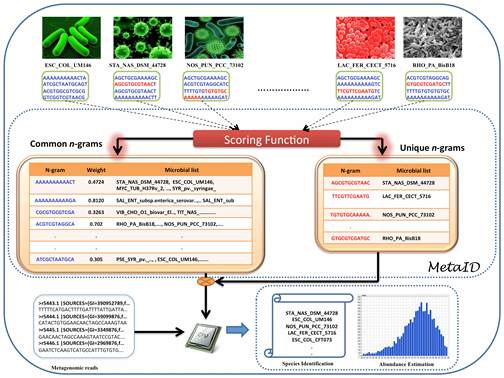 |
| A schematic diagram showing the methodology and scoring function |
Published articles related to this project:
- Srinivasan SM, Guda C. MetaID: A novel method for identification and quantification of metagenomic samples. BMC Genomics (2013). [PubMed]
Graph mining and module detection in protein-protein interaction networks
Protein-protein interaction (PPI) networks carry vital information about proteins’ functions. In this project, we developed graph comparison and graph mining algorithms to detect the subgraphs (that represent a distinct biological functional module in the cell) that are common to all cancers and those that are distinct to each cancer type. This work was conducted in two phases. In the first phase, we constructed nine cancer PPI networks using differentially expressed genes from the Oncomine dataset. From these networks we discovered frequent patterns that occur in all networks and at different size levels. By using effective canonical labeling and adopting weighted adjacency matrices, we are able to perform graph isomorphism test in polynomial running time. Validation of the frequent common patterns using GO semantic similarity showed that the discovered subgraphs scored consistently higher than the randomly generated subgraphs at each size level.
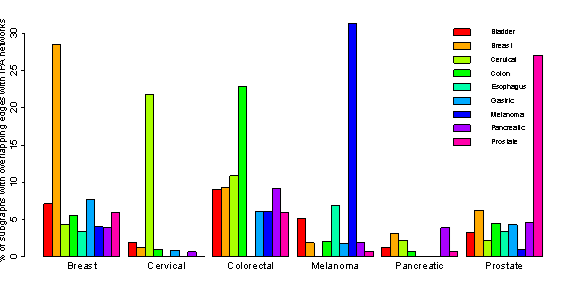
The molecular profiles exhibited in different cancer types are very different; hence, discovering distinct functional modules associated with specific cancer types is very important to understand the distinct functions associated with them. We developed a new graph theory based method to identify distinct functional modules from nine different cancer protein-protein interaction networks. The method is composed of three major steps: (i) extracting modules from protein-protein interaction networks using network clustering algorithms; (ii) identifying distinct subgraphs from the derived modules; and (iii) identifying distinct subgraph patterns from distinct subgraphs. The subgraph patterns were evaluated using experimentally determined cancer specific protein-protein interaction data from the Ingenuity knowledgebase, to identify distinct functional modules that are specific to each cancer type.
Click on image for larger version.
 |
 |
|
Power-law distribution of PPI networks from nine different cancers |
Distribution of distinct subgraphs in PPI networks across the IPA cancer-specific networks |
Published articles related to this project:
- Shen R, Wang, X, Guda C. Mining functional subgraphs from cancer protein-protein interaction networks. BMC Syst Biol. (2012) Suppl 3:S2. doi: 10.1186/1752-0509-6-S3-S2.
- Shen R, Guda C. Applied graph-mining algorithms to study biomolecular interaction networks. BioMed Research International. (2014) 2014:439476. [PubMed]
- Shen R, Gooneshkere NCW, Guda C. Mining functional subgraphs from cancer protein-protein interaction networks. BMC Systems Biology, (2012) 6:S2
Computational methods for predicting protein subcellular localization
This is a long-standing project in our laboratory for over a decade. We have published several computational methods for the prediction of proteins targeted to different subcellular locations using amino acid composition and location-specific domains (MitoPred and pTarget), or based on n-gram based Bayesian classification (ngLOC). We also released an open-source standalone software package to run proteome-scale predictions on local servers. Our ngLOC method was applied to estimate the subcellular proteomes of a number of eukaryotic species. Using the n-gram based classification approach, we developed a new method to identify class-specific subcellular localization signals, some of which are potential novel targeting signals. We have experimentally validated the protein localization predictions from our ngLoc method using GFP-fusion proteins followed by confocal microscopy.
Click on image for larger version.
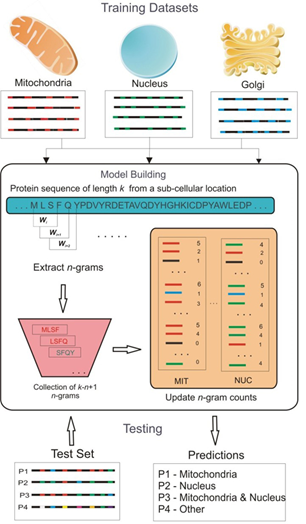 The n-gram model for representing proteins in ngLOC |
Experimental validation of predicted localization of human proteins |
|
An overview of the frequency distribution of amino acids in the signal set for each of the eight subcellular organelles |
|


Published articles related to this project:
- Negi S, Pandey S, Srinivasan SM, Mohammed A, Guda C. LocSigDB: A database of experimental and predicted protein localization signals. Database (Oxford). (2015) [PubMed]
- Chaturvedi NK, Mir RA, Band V, Joshi SS, Guda C. Experimental validation of predicted subcellular localization of human proteins. BMC Research Notes (2014) 7(1):912 [PubMed]
- Srinivasan SM, Vural S, King BR, Guda C. Mining for class-specific motifs in protein sequence classification. BMC Bioinformatics (2013) 14:96.[PubMed]
- King BR, Vural S, Pandey S, Barteau A, Guda C. ngLOC: software and web server for predicting protein subcellular localization in prokaryotes and eukaryotes. BMC Research Notes (2012) 5:351.[PubMed]
- Guda C. Towards cataloguing the subcellular proteomes of eukaryotic organisms. In:Sequence and Genome Analysis (Zhao, Z. ed), iConcepts press. (2010)
- King BR, Guda C. ngLOC:An n-gram based Bayesian method for estimating the subcellular proteomes of eukaryotes. Genome Biology (2007) 8:R68.[Pubmed]
- King BR, Guda C. Semi-supervised learning for classification of protein sequence data. Scientific Programming (2008) 16:5-29.
- King BR, Latham L, Guda C. Estimation of subcellular proteins in bacterial species, The Open and Applied Informatics Journal (2009) 3:1-11. [link]
- Guda C. pTARGET: A web server for predicting protein subcellular localization, Nucleic Acids Research (2006) 35:W210-213. [Pubmed]
- Guda C, Subramaniam S. pTARGET: A new method for predicting protein sub-cellular localization in eukaryotes, Bioinformatics (2005) 21: 3963-3969. [Pubmed]
- Guda P, Subramaniam S, Guda C. MitoProteome: Human heart mitochondrial protein sequence database In: Cardiovascular Proteomics, Methods and Protocols. Methods in Molecular Biology (2006) 357:375-384. [Pubmed]
- Guda C, Guda P, Fahy E, Subramaniam S. MITOPRED: a web server for genome-scale prediction of mitochondrial proteins. Nucleic Acids Research (2004) 32: W372-W374. [Pubmed]
- Guda C, Fahy E, Subramaniam S. MITOPRED: A genome-scale method for prediction of nuclear-encoded mitochondrial proteins. Bioinformatics (2004) 20:1785-1794. [Pubmed]