Rowley Lab
An experimental biology and bioinformatics laboratory discovering principles of 3D genome folding.
The Rowley Lab for 3D Chromatin Organization
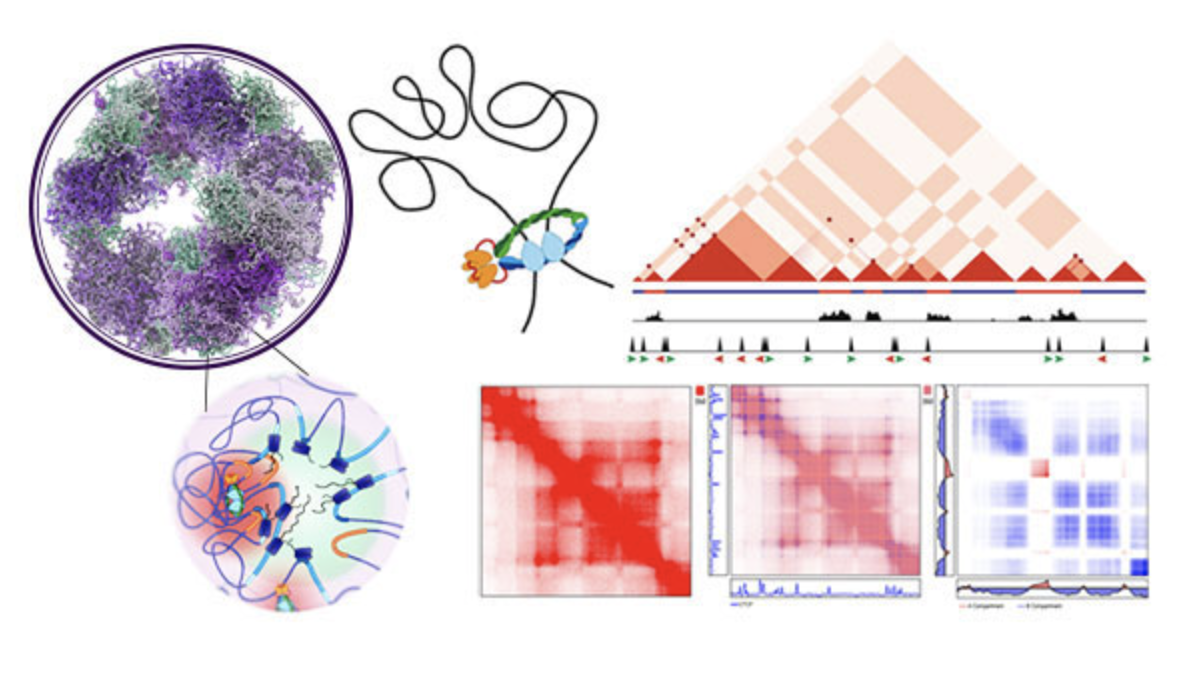
Genomes are intricately folded within the 3D nucleus, providing the context for numerous biological functions. Our research uncovers governing principles of 3D genome folding using a combined bioinformatics and wet-lab experimental approach. We work to better understand the relationships between chromatin, transcription, genome folding, and disease.
Methods
We primarily use genomic methods, including derivations of Hi-C, ATAC-seq, CUT&RUN, and RNA-seq to study features of chromatin organization. We are particularly interested in innovating both the methodology and the analysis that is commonly used to study chromatin architecture in order to understand genome organization at sub-kilobase resolution.
Software Development
Part of our work is to develop novel algorithms and easy to use tools for the analysis of genomics data, particularly data obtained by Hi-C or its derivatives.
Model Systems
We are advocates of using model organisms, which are essential for progress in genetics and have been instrumental in discovering principles of genome organization. We have worked in plants, worms, and flies, each of which has similar and distinct ways of organizing their genome. Understanding these principles is helping to uncover the basic rules of chromatin architecture.Software
Our tools are open-source and versatile. We hope they facilitate your analysis of 3D genome architecture.

CRUSH (coming soon...)
Compartment Refinement for Ultraprecise Stratification of Hi-C (CRUSH) enables sub-kilobase resolution compartment identification from 1/100,000 the reads as well as versatile identification of genome-wide interaction preferences for any feature. Developer: Achyuth Kalluchi
HiCrayon
HiCrayon colors 2D interactions by 1D genomic data. Web app and downloadable versions are available and are particularly useful for striking visuals and comparison of protein occupancy and long-range chromatin interactions. Developer: Ben Nolan
JukeBox (coming soon...)
JukeBox measures noise within a Hi-C map and can be used to evaluate the quality of maps, to predict the return on additional sequencing, or to denote genomic regions of high noise that should be omitted from downstream analysis. Developer: Tim Reznicek
RIPPLE (coming soon...)
Radial Identification of Proximal Propagation within Looped Entities (RIPPLE) identifies the central peak of loop signal and measures the propagation of interactions into proximal loci. Developer: Hannah Harris
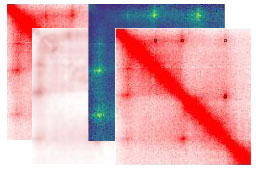
SIP
Significant Interaction Peak caller (SIP) is a tool that uses image processing to identify and analyze loops that appear as high intensity signal in Hi-C maps. Developer: Axel Poulet
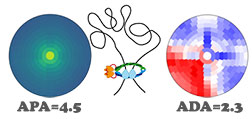
SIPMeta
SIPMeta is used to visualize averages of loops and includes a novel "bullseye" transformation to eliminate visualization biases present in square plots. It also performs aggregate peak analysis (APA) and aggregate domain analysis (ADA). Developer: Axel Poulet

JRowleyLab GitHub
Visit our github page for more! Links to POSSUMM, the NIH/NIGMS Sandbox, and other tools/resources that we have collaborated on or are under development.
Bioinformatics Education
Bioinformatics analysis of 'omics data has become an essential part of biological research. We are committed to educating researchers in analyzing various types of genomic data.
Principal Investigator
Jordan Rowley, PhD
Associate Professor, Department of Genetics, Cell Biology, and Anatomy
Director, Bioinformatics & Systems Biology PhD Program
